TPM와 정규화: raw count를 왜, 어떻게 보정하나
RNA-seq 파이프라인 수업에서 정렬된 read를 유전자마다 세면 raw count가 나오고, 이를 TPM으로 정규화한다고 했습니다. 그런데 왜 정규화가 필요하고, TPM이 정확히 무엇을 어떤 순서로 보정하는지, 그리고 왜 하필 TPM인지, 를 이 문서에서 위젯을 직접 만져 가며 파고듭니다.
한 줄 요약: raw count는 유전자 길이와 시퀀싱 뎁스 두 가지에 오염돼 있어 그대로 비교할 수 없습니다. TPM은 길이 → 뎁스 순으로 보정해, 모든 샘플의 발현량 합계를 정확히 1,000,000으로 고정합니다. 그래서 TPM 값 하나는 “백만 개 전사체 중 이 유전자 몇 개”라는 동일한 잣대가 됩니다.
1. raw count는 두 가지에 휘둘린다
섹션 제목: “1. raw count는 두 가지에 휘둘린다”정렬된 BAM에서 유전자마다 붙은 read를 세면 정수 하나(raw count)가 나옵니다. 문제는 이 숫자가 “그 유전자가 얼마나 발현됐나” 를 곧이곧대로 말해 주지 않는다는 점입니다.
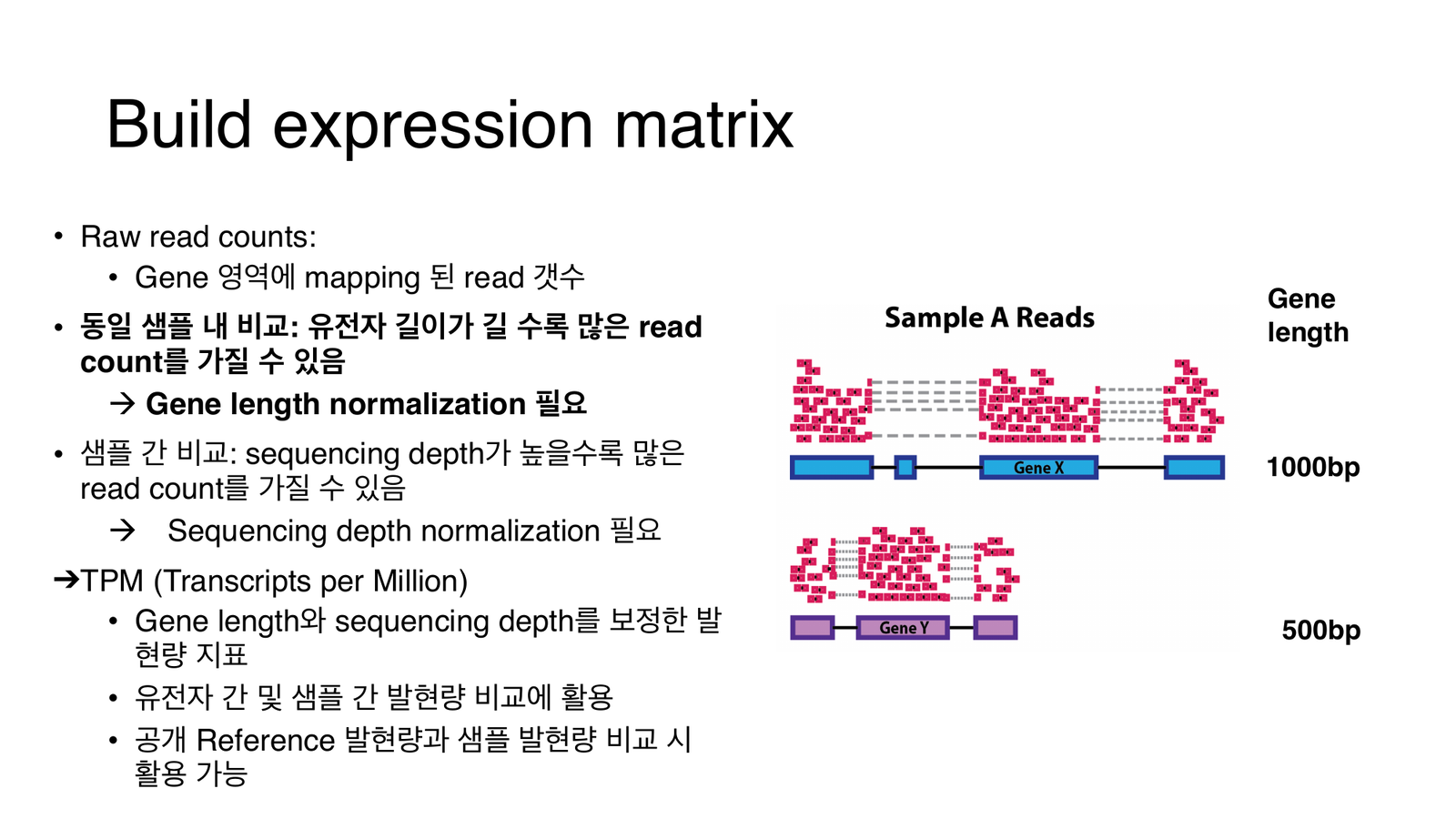
raw count는 두 가지에 휘둘린다: 유전자 길이(길수록 read가 더 붙음)와 시퀀싱 뎁스(많이 읽을수록 전체 count 증가).
함정 ①: 유전자 길이
섹션 제목: “함정 ①: 유전자 길이”mRNA를 잘게 조각내어 시퀀싱하므로, 같은 수의 mRNA 분자라도 긴 유전자에서 조각(fragment)이 더 많이 나옵니다. 즉 read 수는 발현량이 아니라 발현량 × 길이에 비례합니다. 아래 애니메이션에서 유전자 → 전사(mRNA) → 조각내기 → 정렬을 단계별로 따라가 보세요. 발현량(mRNA 3개)을 똑같이 맞췄는데도, 긴 A는 read가 18개·짧은 B는 6개로 벌어집니다. 마지막 단계에서 길이로 나누면 둘이 같아집니다.
이 ”÷ 유전자 길이”가 정규화의 첫 단계이고, 그 결과가 RPK(reads per kilobase) 입니다. 길이가 사라지면 비로소 한 샘플 안에서 유전자끼리 비교할 수 있습니다.
함정 ②: 시퀀싱 뎁스
섹션 제목: “함정 ②: 시퀀싱 뎁스”같은 샘플을 더 깊게 시퀀싱하면 모든 유전자의 count가 그만큼 커집니다. 발현이 변한 게 아니라 더 많이 읽었을 뿐입니다. 그래서 서로 다른 샘플을 비교하려면, 각 유전자 count를 그 샘플의 전체 read 수(뎁스) 로 나눠 스케일을 맞춰야 합니다. 아래에서 같은 조직을 얕게·깊게(3배) 시퀀싱하는 과정을 따라가 보세요: raw count는 전부 3배로 벌어지지만, 전체 read로 나누면 두 샘플이 다시 같아집니다.
정리하면 정규화가 풀어야 할 숙제는 둘입니다: 길이(유전자 간 비교를 막음)와 뎁스(샘플 간 비교를 막음).
2. 한 걸음씩 보정하기: RPK → RPKM/FPKM → TPM
섹션 제목: “2. 한 걸음씩 보정하기: RPK → RPKM/FPKM → TPM”두 보정을 어떤 순서로 적용하느냐가 지표를 가릅니다.
| 지표 | 보정 | 계산 |
|---|---|---|
| CPM (counts per million) | 뎁스만 | count ÷ (전체 read/10⁶) |
| RPK (reads per kilobase) | 길이만 | count ÷ 길이(kb) |
| RPKM / FPKM | 뎁스 → 길이 | count ÷ (전체 read/10⁶) ÷ 길이(kb) |
| TPM | 길이 → 뎁스 | (count ÷ 길이) 를 구한 뒤, 그 합으로 다시 나눔 ×10⁶ |
- RPKM과 FPKM은 사실상 같은 값입니다. read를 세면 RPKM, fragment(paired-end에서 read 쌍 하나 = fragment 하나)를 세면 FPKM일 뿐입니다.
- RPKM/FPKM은 뎁스로 먼저 나눈 뒤 길이로 나눕니다. 그래서 샘플마다 합계가 제각각이 됩니다.
- TPM은 순서를 뒤집습니다. 먼저 길이로 나눠 RPK를 구하고(유전자별 “농도”), 그다음 그 RPK들의 합으로 나눠 백만을 곱합니다. 이 마지막 단계가 결정적입니다: 합으로 나누므로 결과의 총합이 항상 10⁶ 이 됩니다.
3. 지표별 정의와 수식
섹션 제목: “3. 지표별 정의와 수식”이름들이 암호처럼 보이지만, 대부분 “무엇으로 나눴는지”를 그대로 풀어 쓴 약어입니다. 먼저 기호부터:
- : 유전자 에 매핑된 raw read count
- : 유전자 의 길이 (kb 단위)
- : 그 샘플의 전체 매핑 read 수 = 시퀀싱 뎁스
raw count
섹션 제목: “raw count”정렬된 read를 유전자별로 센 원값 . 길이·뎁스 보정이 전혀 없어 그대로는 비교 불가.
RPK: Reads Per Kilobase (킬로베이스당 read 수)
섹션 제목: “RPK: Reads Per Kilobase (킬로베이스당 read 수)”길이(kb)로만 나눕니다.
→ 길이 편향 제거(같은 샘플 안 유전자 간 비교 가능). 뎁스는 그대로. TPM 계산의 중간값입니다.
CPM: Counts Per Million (백만 read당 count)
섹션 제목: “CPM: Counts Per Million (백만 read당 count)”전체 read(뎁스)로만 나누고 을 곱합니다.
→ 뎁스 보정(샘플 간 스케일 정렬). 길이는 그대로라 유전자 간 비교엔 부적합. 함정 ② 애니메이션에서 본 정규화가 바로 이것입니다.
RPKM / FPKM: Reads (Fragments) Per Kilobase per Million
섹션 제목: “RPKM / FPKM: Reads (Fragments) Per Kilobase per Million”“백만 read당·킬로베이스당 read(또는 fragment) 수”. 뎁스 → 길이 순으로 둘 다 나눕니다 (= CPM을 다시 길이로 나눈 것).
- RPKM은 read를, FPKM은 fragment(paired-end에서 read 쌍 하나 = fragment 하나)를 셉니다. paired-end에서 두 read를 한 번만 세면 FPKM: 값은 사실상 같습니다.
- 길이·뎁스 둘 다 보정하지만, 합계 가 샘플마다 달라(→ §5) 샘플 간 비교가 미묘하게 어긋납니다.
TPM: Transcripts Per Million (백만 전사체당 전사체 수)
섹션 제목: “TPM: Transcripts Per Million (백만 전사체당 전사체 수)”길이 → 뎁스 순으로 나누되, 마지막을 자기 샘플의 RPK 합으로 나눕니다.
- 마지막에 합으로 나누므로 항상 . “전사체 100만 개 중 이 유전자 몇 개”라는 일관된 잣대가 됩니다.
- RPKM과의 관계로도 볼 수 있습니다: TPM은 RPKM을 그 합으로 다시 정규화한 값:
💡 이름만 다시 읽어도 정리됩니다: RPK(÷길이) → 여기에 백만당(뎁스)을 붙이면 RPKM, 대신 전사체 비율로 강제하면 TPM. 세 글자 안에 “무엇으로 나눴나”가 다 들어 있습니다.
4. TPM 2단계 계산: 직접 해보기
섹션 제목: “4. TPM 2단계 계산: 직접 해보기”TPM의 실제 계산은 딱 두 단계입니다.
Step 1. 유전자별 count ÷ 길이(kb) = RPK → 긴 유전자일수록 read가 많은 편향 제거 Step 2. 각 RPK ÷ (그 샘플의 RPK 총합) × 10⁶ = TPM → 뎁스 보정 + 총합을 100만으로 고정

작은 예제로 본 TPM 2단계. Step 1에서 길이로 나눠 RPK, Step 2에서 샘플 전체 RPK 합으로 나눈 뒤 스케일링(그림은 설명을 위해 ×10 사용, 실제는 ×10⁶).
아래 계산기에서 지표(Raw/RPKM/TPM) 를 바꿔 가며, 특히 맨 아래 열 합계 Σ 가 어떻게 달라지는지 보세요. Sample A는 네 유전자가 동일하게 발현된 기준 샘플이고, Sample B는 시나리오 버튼·슬라이더로 바꿀 수 있습니다.
5. 왜 TPM이 샘플 간 비교에 유리한가
섹션 제목: “5. 왜 TPM이 샘플 간 비교에 유리한가”위 계산기에서 지표를 TPM으로 두고 Sample B 시나리오를 눌러 보면 한 가지가 드러납니다:
어떤 시나리오에서도 Σ TPM = 1,000,000. 뎁스를 ×3으로 늘려도, 한 유전자를 폭증시켜도 총합은 늘 100만입니다.
이게 핵심입니다. 총합이 고정돼 있으니 TPM 값 하나가 “전체 전사체 100만 개 중 이 유전자의 몫” 이라는 절대적이지 않지만 일관된 잣대가 됩니다. 두 샘플의 TPM을 나란히 놓고 바로 비교할 수 있는 이유죠.
반면 RPKM은 시나리오를 바꿀 때마다 Σ RPKM이 출렁입니다. 합계가 샘플마다 다르면, 같은 RPKM 숫자라도 두 샘플에서 차지하는 비율이 달라, 나란히 비교할 때 미묘하게 어긋납니다.: 이것이 오늘날 발현량 비교에 FPKM보다 TPM을 권장하는 이유입니다.
6. 주의: TPM도 만능은 아니다
섹션 제목: “6. 주의: TPM도 만능은 아니다”TPM이 모든 비교를 해결해 주지는 않습니다. 위 위젯에서 이미 힌트가 나왔습니다.
- TPM은 절대량이 아니라 비율입니다. 총합이 100만으로 고정돼 있어, 한 유전자가 폭증하면(계산기의 ‘G4 폭증’) 다른 유전자들의 TPM이 실제로는 안 변했는데도 떨어져 보입니다. 파이를 나눠 갖기 때문입니다(compositional effect).
- 엄밀한 차등발현(DEG) 분석에는 raw count를 씁니다. DESeq2·edgeR 같은 도구는 “정규화는 우리가 통계 모델 안에서 직접 한다” 며 raw count 입력을 요구합니다. TPM으로 미리 나눠 버리면 그들이 쓰는 정규화(예: median-of-ratios)와 분산 추정이 망가집니다.
- 정리하면 쓰임이 갈립니다: 한 샘플을 레퍼런스 분포와 견주거나(아웃라이어 탐색), 유전자 간·샘플 간 발현을 눈으로 비교할 때는 TPM. 여러 샘플의 통계적 차등발현을 검정할 때는 raw count + 전용 툴.
정리: 지표 한눈 비교
섹션 제목: “정리: 지표 한눈 비교”| 지표 | 길이 보정 | 뎁스 보정 | 샘플 내 유전자 비교 | 샘플 간 비교 | 주 쓰임 |
|---|---|---|---|---|---|
| raw count | ✗ | ✗ | ✗ | ✗ | DEG 도구(DESeq2/edgeR) 입력 |
| CPM | ✗ | ✓ | ✗ | △ | 대략적 뎁스 보정 |
| RPK | ✓ | ✗ | ✓ | ✗ | (중간 단계) |
| RPKM/FPKM | ✓ | ✓ | ✓ | △ (합이 달라짐) | 과거 표준, 지금은 지양 |
| TPM | ✓ | ✓ | ✓ | ✓ (Σ=10⁶) | 발현량 비교·레퍼런스 대조 |
다음 단계가 궁금하다면: 이렇게 만든 TPM 행렬로 한 샘플에서 아웃라이어 유전자를 찾는 과정으로 이어집니다.
- Wagner et al., “Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples”, Theory in Biosciences 2012: DOI
- Lior Pachter, “Models for transcript quantification from RNA-Seq” (TPM 정의): arXiv:1104.3889
- RSEM · featureCounts (Subread) · DESeq2